Pathophysiology of inflammatory rhumatisms
Pathophysiology of inflammatory rhumatisms
1. We address this issue by studying two regulatory molecules furin and adrenomedullin in the pathogenesis of rheumatoid arthritis. Joint destruction in RA results from the influx of immune cells secreting inflammatory cytokines and local depletion of regulatory T lymphocytes (Treg). These cytokines are involved in the metabolism of cartilage and synovial promoting chondrocyte activation / synoviocytaire and catabolism of matrix proteases. We studied two regulatory molecules :
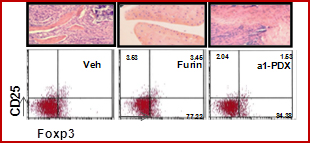
1.1 Furin is a ubiquitous pro protein convertase responsible for the proteolytic cleavage of several cytokines, growth factors and receptors. This study is based on an increased expression and activity of furin in human and murine arthritic joints. Furin could have two different impacts: a catabolic effect by inducing a local excessive maturation of MMP and cytokines and a local and systemic protective effect on the development of Treg. The in vivo approach showed that furin, administered systemically, reduced local inflammation as well as a restoration of the Th1/Th2 balance and the number of Treg. These effects were completely reversed by the inhibitor of Furin, the a 1-PDX (Lin H et al, Arthritis & Rheum 2012). Thus, by promoting the emergence of Treg cells, the furin locally inhibits the development of pannus and MMP expression. These unexpected results have demonstrated a new partner of the mechanisms of immune regulation of RA.
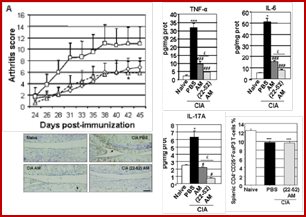
1.2 Another regulatory molecule is adrenomedullin a neuropeptide that is part of the calcitonin, a family of known for antiapoptotic activity, as well as anabolic, angiogenic, proliferative and tumorigenic activity. The production of ADM is increased and the receptors CLR / RAMPs overexpressed in the synovium. The anti-apoptotic role of ADM on rheumatoid synovial fibroblasts is blocked by a peptide derived from the ADM 22-52. ADM stimulates the adhesion of rheumatoid fibroblasts to the extracellular matrix of the synovial tissue, bone and cartilage by activating specific integrin b 1 (Ah-Kioon, Arthritis Res Ther 2011). This effect may be modified in the presence of inflammatory cytokines. In vivo, ADM and its antagonist peptide reduce clinical and histological scores of arthritis, and chondrolysis (Ah-Kioon et al, Arthritis Rheum 2012). This work showed that ADM 22-52 behaved as an agonist of ADM in vivo by expressing higher levels of anti-inflammatory cytokines and anti-apoptotic cells. They modulate both receptors via the ADM expression and secretion of Th1/Th2 balance. Both peptides inhibit IL-17 via the modulation of Treg activity is observed with the peptide derivative. The anti-apoptotic effect of ADM, but especially the peptide, was observed in cartilage in vivo, with a direct or indirect effect via the reduction of local and systemic inflammation.
1.3 Characterisation of immune mechanisms :
RA is characterized by heterogeneity of clinical and biological forms, suggesting the existence of genetic variability in patients. Recently it was shown that the number of variations (CNV) of genes at 22q11.2, a region where the VPREB1 gene encodes the receptor B pre-B cells (pre-BCR), is associated with immune disorders. Since the pre-BCR is involved in negative selection of autoreactive B cells, we postulated that the CNV in the gene VPREB1 may be responsible for the loss of B cell tolerance in RA. In collaboration with Chung Hun-Tag (University of Ulsan, South Korea), we showed the existence of an association between low PREB1 CNV gene and RA. We also observed a decrease in immature B cells / transitional and increased memory B cells in patients (Yim et al. Mol Immunol 2011). Our results suggest that the CNV abnormalities observed may contribute to negative selection mediated by the pre-B in RA. In parallel, we studied the involvement of chemokines and their receptors in the recruitment of inflammatory cells and in angiogenesis associated with RA and other inflammatory rheumatic diseases. Previously we showed that superantigens (SAGs) for B cells interact with the hypervariable regions of immunoglobulins (Igs) can impact human and 30-50% of circulating human B cells and induce their proliferation or death by apoptosis. M. Zouali found that a SAg is capable of modulating the expression of the chemotactic receptor CXCR4 and inhibit chemotaxis of human B lymphocytes to ligand CXCR12. These effects are temperature-dependent and persisted for more than 24 hours after treatment with the SAg. This preferentially targets naive B lymphocytes, and decreases the expression of sIgM and sIgD, without affecting that of CD19. Indeed, confocal microscopy experiments and cell biology have shown that SAg binds to the sIgM / D on the surface of B cells, and induces its internalization (Bohris et al. Mol Med, 2012). These observations may have therapeutic implications. Indeed, the CXCR4 receptor is associated with certain autoimmune diseases. Since the SAg acts as an inhibitor of CXCR4 and chemotaxis of human B cell, and given that blocking CXCR4 has a beneficial effect on disease progression, these results suggest that inhibition of CXCR4 by the Sag may have therapeutic value in autoimmune diseases.